
Disclaimer:
The content on this website is for informational and educational purposes only and is intended to help readers understand AI technologies used in healthcare settings. It does not provide medical advice, diagnosis, treatment, or clinical guidance. Any medical decisions must be made by qualified healthcare professionals. AI models, tools, or workflows described here are assistive technologies, not substitutes for professional medical judgment. Deployment of any AI system in real clinical environments requires institutional approval, regulatory and legal review, data privacy compliance (e.g., HIPAA/GDPR), and oversight by licensed medical personnel. DR7.ai and its authors assume no responsibility for actions taken based on this content.
This is a personal/prototype-level experience, not a clinical protocol.
Medical AI regulations aren’t a roadblock, they’re the specification for safe, shippable systems. I have experience navigating validation processes in hospital research settings and have dealt with the “can we deploy this by Q4?” pressure. In this guide, I translate the United States, EU, and key global frameworks into concrete steps so you can design for clearance from day one, collect the right evidence, and keep post-market drift under control.
Table of Contents
United States: FDA Oversight of Medical AI and Machine Learning
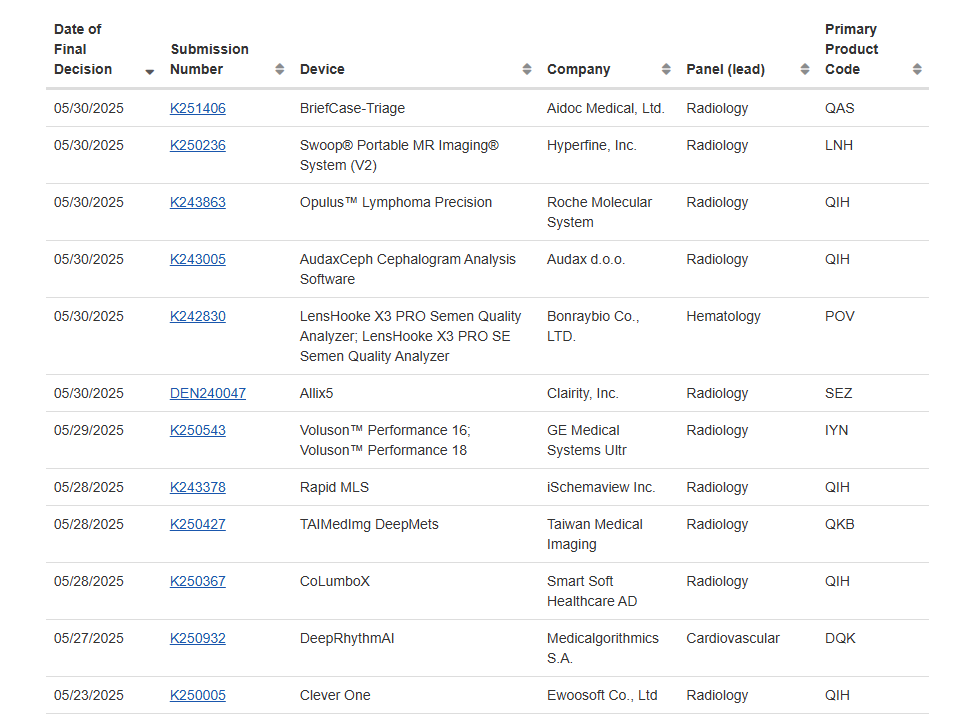
How the FDA Regulates AI/ML as Medical Devices (SaMD)
If your model informs diagnosis, treatment, or clinical workflow decisions, the FDA will likely treat it as Software as a Medical Device (SaMD). The agency maps AI/ML SaMD to existing device classes based on risk and intended use, then applies quality system and evidence expectations accordingly. Key anchors:
- SaMD foundations: IMDRF principles adapted by FDA and reflected across guidances (see FDA’s “Good Machine Learning Practice” discussion papers and device listings as of 2025).
- AI/ML-enabled device inventory: FDA’s official AI/ML-enabled medical devices list shows precedent indications and evidence patterns for exemplars of radiology CAD, triage, and decision support.
- GxP expectations: Even without a finalized AI-specific regulation, FDA leans on 21 CFR 820 (Quality System Regulation: transitioning to QMSR alignment with ISO 13485), cybersecurity premarket guidance (2023–2024), and human factors principles for clinical UI.
FDA Clearance Pathways and Compliance Guidelines for AI in Healthcare
I plan US submissions around three lanes:
- 510(k): For substantial equivalence to a predicate (common for imaging CAD). Your validation must show non-inferiority and clinical relevance in representative data.
- De Novo: For novel, moderate-risk indications when no predicate exists. Heavier benefit-risk justification: still achievable with strong clinical evidence and risk controls.
- PMA: For high-risk indications (e.g., autonomous diagnosis) requiring robust pivotal studies.

What I build into the technical file:
- Intended Use and Claims: Narrow, testable, and clinically meaningful. Avoid “catch-all” claims that inflate evidence needs.
- Data governance: Prospectively defined datasets: demographic and site diversity: shift analysis: PHI handling under HIPAA.
- Model performance: Primary endpoints that map to clinical benefit (AUROC is not enough). Include sensitivity/specificity at clinically set thresholds, reader study impact, and failure mode analysis.
- Change management: Predetermine what can change post-clearance. FDA has outlined a “Predetermined Change Control Plan” concept for AI/ML, specify retraining triggers, guardrails, and verification/validation (see FDA discussion in GMLP papers and 2024–2025 draft guidances).
- Risk management: ISO 14971-aligned hazard analysis: mitigations spanning UI, monitoring, and rollback. Reference FDA’s benefit-risk frameworks and cybersecurity controls (SBOMs, secure update channels).
References: FDA’s Good Machine Learning Practice guidelines and FDA’s clinical decision support software guidance provide essential frameworks for AI/ML device development.
European Union: CE Marking and MDR Requirements for AI in Medicine
How EU MDR Applies to Medical AI Systems
Under EU MDR (Regulation 2017/745), clinical decision-making software that drives diagnosis/treatment is a medical device, often Class IIa or IIb (potentially III for high-risk autonomous functions). MDR emphasizes:
- Clinical evaluation: Demonstrate clinical performance and clinical benefit with systematic literature, bench/analytical validation, and clinical investigations where needed.

- PMS/PMCF: Post-market surveillance and post-market clinical follow-up are not optional, expect real-world evidence and vigilance reporting.
- QMS: ISO 13485-backed QMS with software lifecycle per IEC 62304, usability (62366), and risk (14971).
CE Marking Process and Conformity Assessments for AI/ML Tools
My CE playbook:
- Classify: Justify classification using MDCG guidance: define intended purpose precisely.
- Technical documentation: State of the art: data representativeness: performance metrics: bias analysis: cybersecurity: algorithm change plan.
- Notified Body assessment: Most AI will require NB review. Build a clear clinical evaluation report (CER) with traceability from claims to evidence.
- AI Act interface: EU AI Act (Regulation 2024/1689) overlays MDR with transparency, data governance, and risk controls for “high-risk” AI used in healthcare. Align data provenance, logging, and human oversight with AI Act articles to avoid rework.
References: WHO’s Ethics and Governance of AI for Health guidance (2023) provides useful governance patterns for fairness and transparency.
Global Medical AI Regulations Beyond the US and EU
Regulatory Approaches in the UK, Canada, and Other Leading Countries
- UK: MHRA’s Software and AI as a Medical Device Change Programme sets out evidence and lifecycle expectations aligning with SaMD and AI adaptivity, with upcoming UKCA pathways. Converges on risk-based claims, real-world monitoring, and change management.
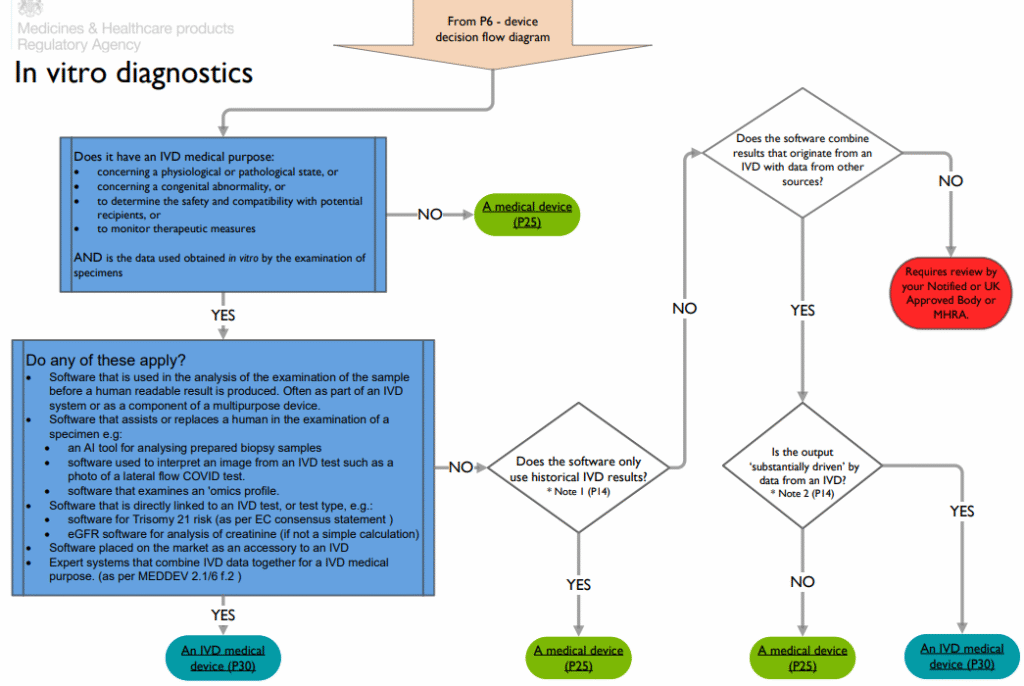
- Canada: Health Canada classifies SaMD via risk and requires evidence proportional to claims: it recognizes MDSAP and leans on IEC 62304, 14971, and cybersecurity guidance.
- Others (Australia, Singapore): Generally IMDRF-aligned with clear SaMD rules and growing AI guidance.
Emerging AI Healthcare Frameworks in China, Japan, and APAC
- Japan: PMDA applies device classifications with rigorous clinical evaluation: software typically follows JIS/IEC harmonized standards.
- China: NMPA requires detailed algorithm documentation, dataset provenance (local data often favored), and cybersecurity filings.
- Singapore/ASEAN: MAS/IMDA and HSA guidance emphasize transparency, audit trails, and post-market monitoring for AI-based CDS.
WHO’s 2023 guidance on AI in health stresses safety, effectiveness, equity, and accountability, good scaffolding for any market.
Cross-Regional Compliance Themes in Medical AI Regulations
Risk Management, Clinical Validation, and Model Performance Requirements
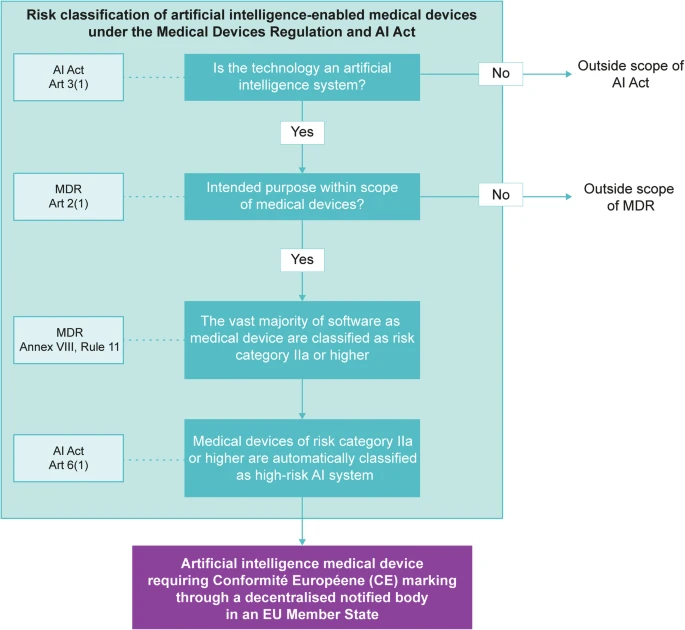
Across regions, I see the same non-negotiables:
- Risk-first design (ISO 14971) with clear hazard controls.
- Endpoints tied to clinical benefit: reader time-to-diagnosis, sensitivity at action thresholds, net reclassification improvement, workflow impact.
- External validation: Multi-site, demographically diverse data: prespecified analysis: calibration and subgroup performance.
- Hallucination and brittleness metrics for LLMs: hallucination rate under retrieval-augmented prompts, answer abstention rate, factuality against a gold corpus, and post-deployment drift KPIs.
Documentation, Transparency, and Algorithm Change Management
- Traceability: Map every claim to evidence, tests to requirements, and risks to mitigations.
- Transparency: Explainable intended use, training data characteristics, and known failure modes.
- Change control: Define retraining triggers, verification/validation steps, and rollback plans. FDA’s Predetermined Change Control Plan, EU MDR’s PMS/PMCF, and UK MHRA’s change programme all converge on documented, monitored adaptivity.
Reference points: FDA device listings and GMLP papers: WHO AI in Health 2023: MDR and AI Act texts: MHRA SaMD/AI programme.
How Developers Can Prepare for Medical AI Regulatory Approval
Practical Steps to Ensure Regulatory Compliance from Early Development
What I build into sprint zero:
- Specs first: Freeze intended use, indications, and user population. Write claims as testable requirements.
- Data plan: Source representative, consented data: document lineage: set aside locked external test sets: pre-register analysis where feasible.
- Benchmarks: Beyond AUROC, define thresholded clinical metrics, calibration, time-to-decision, and human-in-the-loop outcomes. For LLMs, track hallucination rate, harmful content, PHI leakage, and refusal behavior.
- Risk files: Start ISO 14971 hazard analysis early: include misuse scenarios and human factors.
- MLOps/QMS: Version everything (datasets, code, model cards): automate validation pipelines: generate audit-ready logs and SBOMs.
- Security & privacy: HIPAA/GDPR-compliant data handling: threat modeling: encryption and access controls.
- Pre-sub engagement: In the US, use Q-Sub to get FDA feedback on claims, study design, and PCCP. Similar early dialogues help in EU/UK.
When and How to Work With Regulatory Experts for AI Healthcare Projects
Bring in regulatory counsel once claims and architecture stabilize but before clinical studies. I pair experts with engineering to:
- Stress-test indications and classification.
- Right-size evidence (bench, reader study, clinical investigation).
- Draft the Predetermined Change Control Plan and PMS strategy.
- Align documentation with FDA guidances (145022, 181442, 184150), EU MDR/AI Act, and MHRA SaMD programme.
Ethics, safety, and emergency caveats: This article is informational and not medical advice. Do not use AI outputs for diagnosis or treatment without licensed clinician oversight. Seek emergency care for acute symptoms. Always consult qualified healthcare professionals and your regulatory team before deployment.
Disclosures and currency: I have no financial ties to vendors mentioned. Content reflects regulations and public guidance as of November 2025. Key sources: FDA AI/ML device listings and guidances: WHO 2023 AI in health: EU MDR (2017/745) and AI Act (2024/1689): MHRA SaMD/AI change programme: and recent peer-reviewed literature on clinical AI evaluation.
Past Review:





